How real-world evidence supports oncology drug development
RWE in oncology must stand up to regulatory, HTA, and access scrutiny—requiring decision‑grade, multimodal, globally relevant data.

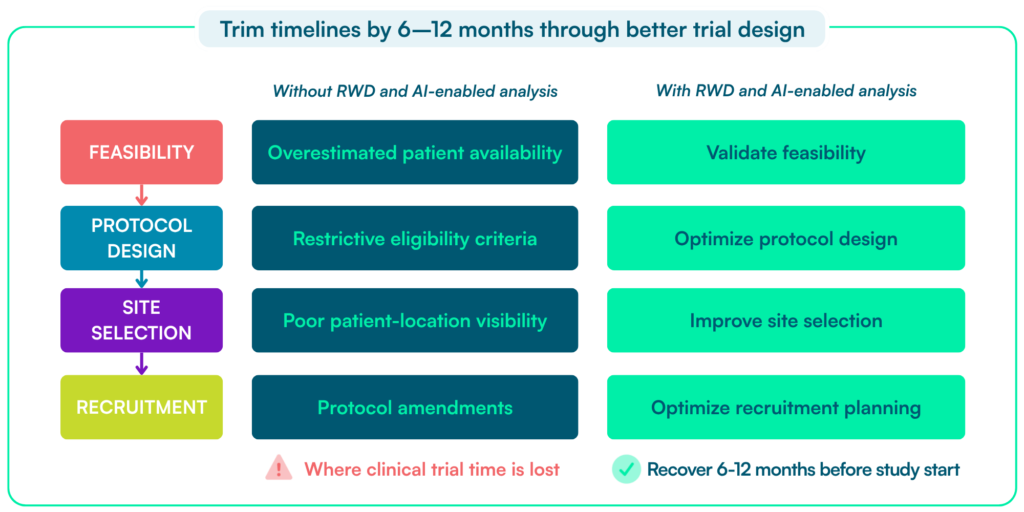
Pharma often focuses on execution. Teams open more sites, push enrollment harder, and try to recover time once delays appear. The bigger opportunity sits earlier. Most timeline risks are built into the study during feasibility, protocol design, and site strategy. These decisions often rely on incomplete data. Every month lost has real impact: patients wait longer, teams spend time fixing avoidable issues, revenue shifts further out.
The shift is straightforward. Instead of recovering time later, teams can prevent delays early by testing assumptions before finalizing the protocol. That means answering key questions early:
Fast trials start with better design.
For a deeper look at clinical trials and how early design decisions shape risk, read How real-world data helps reduce clinical trial risk.
In practice, delays come from four areas:
These are not operational failures. They result from early decisions made with incomplete data. Their impact compounds across the study: recruitment slows, amendments increase, and teams spend months correcting assumptions.
AI helps teams test these assumptions earlier by evaluating feasibility, protocol, and recruitment scenarios before execution.
Early planning decisions matter. Clinical trial delays are measured in months, carry significant financial impact, and often stem from the same root causes: unrealistic feasibility assumptions, recruitment shortfalls, and protocol changes.
A typical trial runs nearly a year longer than planned. This delays regulatory submission and market entry.
Under-enrolled trials risk being statistically underpowered. This can lead to inconclusive results or additional studies.
They remain one of the primary drivers of timeline failure across clinical development.
Common amendments include updating eligibility criteria, adding or removing endpoints, adjusting visit schedules, and expanding study sites or countries. Each change requires regulatory review, ethics approval, site retraining, and system updates. This disrupts ongoing recruitment and execution.
Even smaller changes typically add months. For example, modifying eligibility criteria requires pausing recruitment, securing approvals, retraining sites, and re‑screening patients before enrollment can resume.
The financial impact of delay is measurable in two ways⁵:
Recent Tufts CSDD research estimates that each day of delay represents approximately $800,000 in delayed or lost prescription drug or biologic sales. The direct daily cost of conducting a clinical trial averages about $40,000. The first figure reflects delayed revenue opportunity from later market entry. The second reflects the ongoing operating cost of keeping a trial running.
For a six‑ to twelve‑month delay, the commercial impact can reach tens to hundreds of millions of dollars. This depends on the product, indication, launch assumptions, and trial profile. This should be understood as delayed or unrealized revenue opportunity—not as an out‑of‑pocket trial expense.
Most delays start before or shortly after a study begins—during feasibility, protocol design, and early recruitment planning. Once assumptions are set, they are hard to fix. The opportunity to recover 6–12 months comes from fixing these issues early — before the protocol is finalized, sites are selected, and recruitment assumptions are set.
Recruitment is often treated as the primary bottleneck, but the underlying issue starts earlier. By the time a study is “behind on enrollment”:
Recruitment failures are rarely random. They usually reflect avoidable decisions made months before the first patient is screened—in feasibility planning, protocol design, eligibility criteria, and site selection. Each additional eligibility criterion reduces the pool of patients who can realistically be enrolled, often more than teams expect during protocol design. Reactive fixes—adding sites, expanding geographies, or loosening criteria—rarely recover lost time.
At that stage, the delay is already embedded in the design.
Feasibility and study design decisions have traditionally relied on site feasibility assessments. Investigators estimate how many patients they can recruit and how quickly.
Real-world data (RWD) takes a different approach. It uses observed (and anonymized) patient-level information—who is diagnosed, treated, and eligible in routine clinical practice, and where.
AI builds on this by enabling teams to analyee these populations at scale. It allows teams to quantify eligibility, test protocol assumptions, and evaluate feasibility scenarios much faster than traditional approaches.
This matters because feasibility estimates are often unreliable. Teams typically select sites based on feasibility questionnaires and projected recruitment numbers. Studies show that teams often overestimate feasibility and that many trials miss their recruitment targets¹.
RWD addresses this gap. It grounds decisions in actual patient populations rather than estimates. Instead of projecting availability, teams can quantify how many patients meet the study criteria across regions and treatment settings. They can test how eligibility criteria affect the size of the recruitable population and identify where those patients are treated.
RWD also helps ensure that study design reflects clinical reality. Teams can evaluate visit frequency, procedures, and follow-up schedules against routine care pathways. This reduces patient burden and increases the likelihood that eligible patients can be recruited and retained.
Better data does not speed up recruitment once a study is underway. It prevents studies from entering execution with unrealistic assumptions about patient availability, eligibility, and site performance. This reduces the need for amendments and lowers the risk of delays.
Once these data foundations are in place, AI helps teams test assumptions faster. But it cannot compensate for poor or incomplete data.
AI does not replace clinical judgment — it accelerates the number of decisions teams can test before committing to a study design. AI supports clinical trial planning by enabling faster analysis of patient populations, protocol scenarios, and feasibility assumptions. Its impact depends on the underlying data.
As our Global Head of Technology and Data Services, Narasimha Kumar puts it:
AI readiness is fundamentally about evidence (data) readiness.
AI Readiness in Pharma – Getting the Foundation Right, Bio‑IT World
AI helps scan large datasets, identify patient cohorts, compare protocol scenarios, and surface patterns that manual review may miss. When data is curated and longitudinal, these capabilities shorten planning cycles and support better trade-off decisions.
This matters most in early study planning, where delays often result from repeated rounds of alignment.
Clinical, medical, biostatistics, and operations teams must iterate on sample size assumptions, endpoints, eligibility criteria, and country strategy. AI accelerates these “what‑if” analyses. It makes discussions more evidence-based and reduces reliance on slow manual iteration.
But AI has limits:
Data quality is the limiting factor. When clinical, genomic, and real-world data are incomplete, fragmented, or inaccessible, AI-driven analyses reflect those same gaps. This leads to unreliable feasibility and design decisions.
When data is robust and well integrated, AI helps teams evaluate assumptions faster and reach better decisions earlier.
With the right data, teams can use AI to assess study feasibility before execution:
AI does not make sites recruit faster. It reduces the risk of designing a study that cannot recruit as planned.
One of the most important design risks is whether the trial population reflects the patients who actually exist in routine care.
Speed without representativeness creates risk. A trial may progress quickly but still fail if it does not reflect the intended patient population. This can happen through slow recruitment, protocol changes, or limited regulatory confidence.
Regulatory expectations are explicit. The FDA recommends that trials enroll populations representative of the patients who will use the drug. This includes variation in demographics, comorbidities, and clinical characteristics³.
This reflects a broader shift: trial design must align with real-world patient populations from the outset. The implications are operational, not just scientific.
When populations are not representative, recruitment assumptions break down. Patient availability is often overestimated during planning.
This is a well-documented phenomenon known as Lasagna’s law. The number of patients who can actually be enrolled is often far lower than initial estimates⁴. This directly leads to slower enrollment and missed timelines.
In practice, teams often need corrective action. Studies may require changes to eligibility criteria, site strategy, or study design after initiation. These adjustments add complexity and delay that earlier planning could have avoided.
Non-representative populations also weaken confidence in results. If study participants do not reflect real-world patients, regulators and clinicians may question whether findings apply in practice. This reduces the value of the evidence generated³.
These issues are closely linked. They stem from the same root cause: limited visibility into real patient populations during feasibility and study design.
Representative RWD addresses this earlier in the process. It allows teams to quantify who is actually reachable for a study, test how eligibility criteria affect the recruitable population, and align site strategy with where those patients are treated.
The result is not just stronger evidence. It leads to more realistic recruitment assumptions, fewer late-stage adjustments, and more predictable timelines.
Better data changes the timeline equation. It helps teams resolve feasibility, eligibility, site strategy, and recruitment assumptions before they become operational problems.
Individually, these delays may seem manageable. Together, they often extend timelines by six to twelve months or more. Better data helps prevent these delays before they occur.
Avoiding six to twelve months of delay does more than improve operational efficiency. It also protects commercial value by reducing delayed revenue opportunity and unnecessary trial operating costs.
When teams quantify patient populations accurately, test eligibility criteria against real-world data, and align site strategy with where patients are treated, studies are more likely to meet recruitment assumptions and proceed without major changes.
AI accelerates this process by reducing the time needed to test scenarios and align decisions. It does not change the underlying outcome. The key is that teams make decisions on realistic, data-driven assumptions from the start.
The result is fewer amendments, fewer corrective actions, and more predictable execution—recovering months that would otherwise be lost during the study.
AI supports this by enabling faster iteration. Teams can test and refine assumptions before they are locked into study design.
Clinical trial timelines are not primarily extended by slow execution. They are extended by early decisions made with too little data.
Improving visibility into real patient populations allows teams to resolve feasibility and design issues before they turn into recruitment delays and protocol changes.
For organizations aiming to shorten development timelines by six to twelve months, the opportunity is clear: focus less on accelerating execution and more on getting the design right from the start.
BC Platforms supports this by providing access to harmonized clinical, real-world, and genomic data across regions.
Teams can quantify patient populations, test eligibility criteria, and assess feasibility across geographies before the study begins.
Combined with AI-driven analysis, teams can run multiple feasibility scenarios and align decisions before execution.
This allows teams to validate study assumptions early, rather than correct them later through amendments and recruitment adjustments. The result is fewer feasibility failures, fewer protocol changes, and more predictable timelines.
See how real-world data can help validate patient availability, refine protocol design, reduce amendments, and improve recruitment planning.
Real‑world data improves planning by replacing assumptions with observed patient populations. Teams can quantify eligible patients, test criteria, and identify where patients are treated—reducing recruitment risk before the study starts.
AI accelerates feasibility and protocol design by enabling faster scenario testing. Teams can evaluate patient populations, test eligibility criteria, and compare design options more quickly than with manual analysis.
Representative data ensures recruitment assumptions reflect real patient populations. This reduces the risk of slow enrollment, protocol changes, and delays later in the study.
BC Platforms helps shorten timelines by enabling early validation of feasibility and study design. With access to harmonized clinical, genomic, and real‑world data, teams can test assumptions early and reduce recruitment risk and protocol amendments.